Research
Theoretical Chemical Biology and Protein Modelling
The driving force behind our research is to gain a deeper understanding of the dynamics and specificity of biological processes, like molecular recognition or enzymatic reactions. For this purpose, we develop novel computational methods, which combine approaches from computational biochemistry, bioinformatics, theoretical biophysics, and statistics. Next to the methodological projects we apply our and other methods in computational studies of specific biochemical systems, which are performed in close collaboration with experimental groups, predominantly in the areas of computer-aided drug design, protein and enzyme engineering, and computational immunology.
One of the major bottlenecks for the computational description of biological processes is the limitation in the time scale and system size, which can be covered by the existing theoretical methods. Much research has been devoted to this problem and many advanced biophysical methods have been developed for this task. Most of them are, however, very time consuming and cannot be used for applications in which many different situations must be investigated simultaneously, as in protein or drug design. To be able to deal with such applications, we develop hierarchical models, which combine very efficient, discrete sequence- and structure-based methods from bioinformatics with more demanding continuous biophysical approaches, such that it is possible to always use the most efficient method for the application at hand. All methods are implemented in a new software package, DynaCell.
Our methodological work in the context of computer-aided drug design focuses on the development of efficient sampling and scoring algorithms for protein-ligand assembly and the application of our methodology in practical drug design projects with our experimental partners. In addition to our research in computer-aided drug design we are working on topics concerning the areas of protein/enzyme engineering and structural immunoinformatics. In these areas we develop efficient methods for protein mutation, structural modeling of immunologically relevant proteins like T-cell receptors and antibodies, and the combined sequence- and structure-based prediction of MHC class I/II epitopes. In addition, we investigate the effect of mutations on the function and stability of different proteins and their complexes.
Specific research areas:
Efficient Treatment of Protein Flexibility in Drug and Protein Design
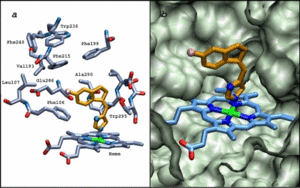
In molecular docking and protein structure prediction traditionally discrete optimization approaches are applied. This, however, is a very crude approximation of the real dynamics of such systems and for many processes and systems not appropriate. By combining discrete (IRECS) and continuous, biophysics-based (DynaDock) approaches we try to extend the area of applicability of in silico methods in these fields. With IRECS it is e.g. possible to predict the conformations and the flexibility of side chains very efficiently. Afterwards the IRECS-based protein structures can be further optimized by the program DynaDock, which allows for a very efficient molecular dynamics-based treatment of backbone flexibility. Both methods are among the best performing approaches for studying and predicting protein-peptide interactions, side chain placement, and docking into homology modeled protein structures and flexible binding sites.
Development of new Energy and Scoring Functions
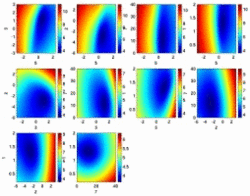
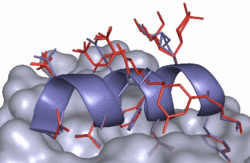
In molecular modeling the simulation/docking algorithm used and the corresponding scoring/energy function are closely related. Therefore the development of new sampling algorithms must be accompanied by the design of the appropriate scoring functions. Together with our sampling and simulation algorithms, we develop energy and scoring functions for an efficient, yet accurate prediction of protein-ligand binding (ROTA, pepscore, BiPPred). In addition, we are designing automated approaches for a system-specific optimization of different energy functions (POEM).
Computer-aided Drug Design Applications
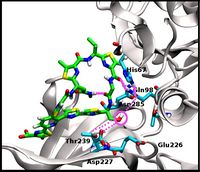
Next to the methodological developments, we also apply the above methods and others in application studies in the area of computer-aided drug design, which are performed in close collaboration with experimental groups. In this context we study inhibitor and substrate binding to various proteins, like CYP P450 enzymes, Hsp70 chaperones, Methyl- and Glycosyltransferases, elongation factor EF-Tu, Protein Disulfide isomerase, different proteases (e.g. HCV NS3-4A protease, Caseinolytic Protease P), and trans-membrane transporters.
Computational Immunology
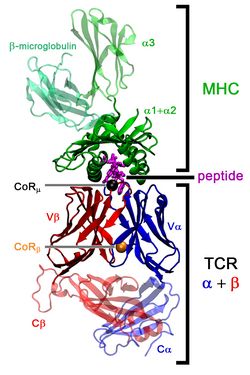
Computational Immunology is a new, fast growing area at the borderline of biophysics and bioinformatics. Until recently the field was dominated by sequence-based prediction approaches, mainly due to the lack of sufficient structural information. However, the amount of structure-based data has been growing fast during the last years and thus structural approaches are now feasible.
Our research in this area focuses on the combined sequence- and structure-based prediction of T-cell response, specifically T-cell receptor-peptide-MHC binding, and on the development of accurate structural approaches for T-cell receptor and antibody modeling. We entered the field by developing a new prediction method, DynaPred, for the prediction of the binding of endogenous antigenic peptides to MHC class I molecules and performing docking studies of medically relevant peptides to MHC molecules with IRECS and DynaDock. Currently we are focusing on the design of efficient structural modeling approaches for T-cell receptor-p-MHC complexes and antibodies (MoFvAb and DynaDom).
Enzyme and Protein Engineering and Medical Applications
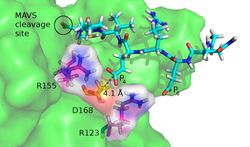
Next to our research in computer-aided drug design and computational immunology we are active in the areas of protein and enzyme engineering. In this context, we predominantly study the effect of mutations on the function and stability of different proteins as mutations can have a major effect on the substrate and inhibitor binding affinity as well as the stability of the corresponding protein. Thus, the prediction of these effects is very important in various fields from medicine to bioengineering. For this purpose we developed a special side chain placement tool IRECS, which can additionally predict side chain flexibility and is thus also very useful for computer-aided drug design studies. Using IRECS in combination with biophysical methods like e.g. molecular dynamics simulations we studied the effect of binding site mutations on ligand binding in various application projects, e.g. investigating the effect of drug resistance mutations on the stability and dynamics of the Hepatitis C viral NS3-4A protease.